From a machine learning professional’s perspective, the drug development process is essentially an optimisation task. It requires a team of scientists and professionals, takes 10 to 15 years and costs up to 2 billion euros (including the costs of unsuccessful projects). The process is a sequence of steps where researchers start with thousands of compounds and optimise down to a few successful candidates. To understand where machine learning is helpful, we first need to break down drug development into parts. [1]
Blog

Machine learning in drug discovery
Maria Yli-Luukko Senior Data Scientist, Solita
Published 18 May 2026
Reading time 7 min

A common assumption is that generative models make predictive models less relevant. The opposite is closer to the truth. A generator that produces billions of novel structures is only as useful as the filters downstream, and those filters are often predictive models. This post goes through the common drug discovery pipeline, the role of machine learning in it, and a closer look at molecular property prediction as a component.
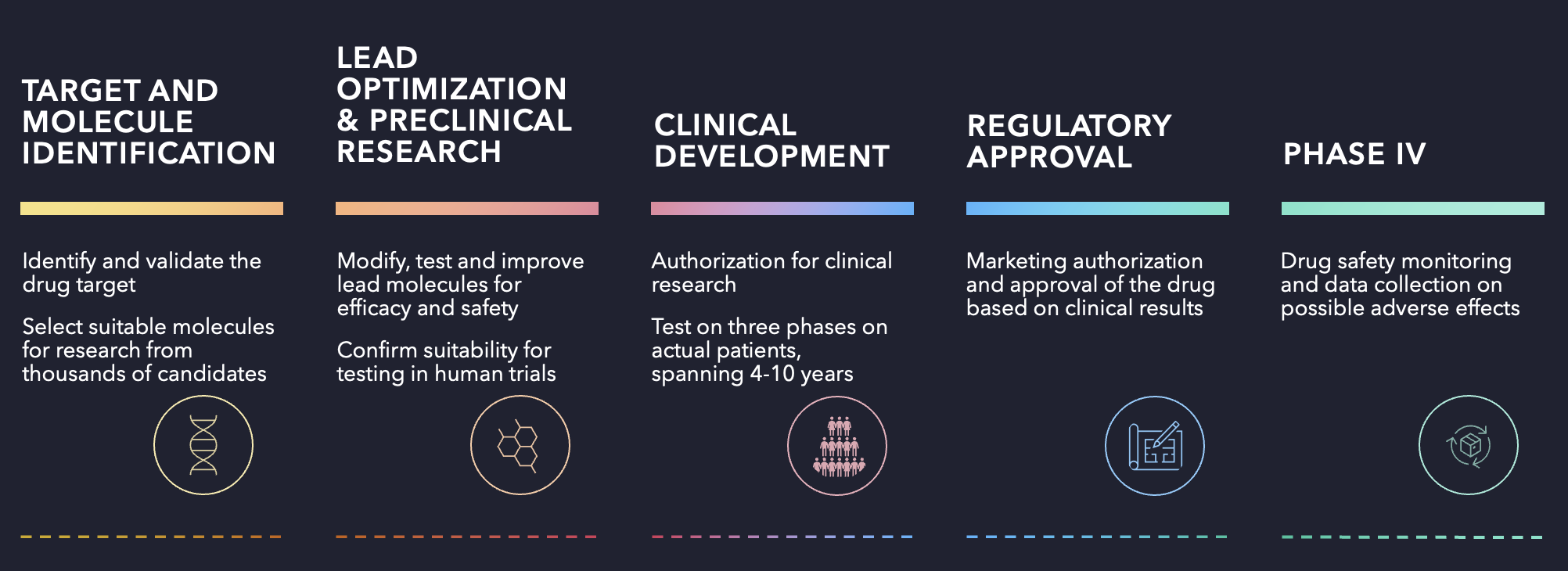
1. Target and molecule identification and validation
Drug development starts by identifying a drug target in the body and demonstrating experimentally that it produces a therapeutic effect on a given symptom or disease. After the target is validated, screening campaigns evaluate up to thousands of compounds to narrow down to the most promising candidates for further study. [1][2]
2. Lead optimisation and preclinical research
The selected leads are then tested with the goal of reducing them to a small set of molecules with the best balance of efficacy and safety. Those drug candidates move into preclinical research, where they must demonstrate that they are safe enough to be administered to humans.
3. Clinical development
Human trials follow, requiring approval from authorities. At this point, the research has taken 3-5 years.
The trials proceed in three phases:
- Phase I (~1 year): 20–100 healthy volunteers receive the drug as part of clinical trials. Roughly a tenth of molecules that enter this phase become a finished product.
- Phase II (1–3 years): The drug is given to actual patients (100–500) for the first time, to verify that the effect is reproducible and to gather more data on behaviour and safety.
- Phase III (2–4 years): A statistically significant group of 1000–5000 patients across hundreds of clinical sites worldwide is used to establish efficacy and safety even further.
4. Regulatory approval and phase IV
If phase III delivers, the marketing authorisation application is filed, and review by the regulator takes around a year. Once the drug reaches patients, phase IV (post-marketing surveillance) begins and continues for the drug’s entire lifetime, with adverse-event and usage data collected on an ongoing basis.
Where and how can ML be used?
The chapter above describes the drug development process in rough terms: the process itself has multiple steps, and a significant number of candidates fail during the development. Machine learning models can be used to reduce the amount of time and costs spent on bad candidates by filtering them out early, as well as creating novel ideas for new candidates. Here are a few examples of virtual ML use cases in drug development:
- Generating new compounds: Using generative algorithms to design novel molecular structures based on specific intents or constraints.
- Virtual screening: Predict the activity of a drug candidate based on existing observations and screen large databases of compounds for potential drug candidates (HTS).
- Target identification: Analysing libraries and datasets to discover new disease-modifying targets and potential drug candidates.
- Molecular property prediction: Estimating how a compound will behave biologically and chemically before it is synthesised (we will focus on this below).
- Automated document review: Using Natural Language Processing (NLP) to parse through regulatory documents and scientific literature.
- Early signal detection: Monitoring real-world data to identify adverse events, polypharmacy side effects, and food-drug interactions long after a drug has reached the market. [3][4]
Molecular property prediction
We focus on one of the approaches above: molecular property prediction. Before a machine learning model can predict a molecule’s properties, it must be translated into a mathematical format that the computer can read and understand. This process is known as featurisation.
There are several standard ways to featurise small molecules, and to name a few:
- SMILES 1D representation: A string of characters that represents a molecule’s atomic structure. Language models can read SMILES strings much like they read text.
- Fingerprints: A bit-vector array that indicates the presence or absence of specific molecular substructures.
- Graphs: Representing atoms as nodes and chemical bonds as edges, which allows using Graph Neural Networks (GNNs) to capture the molecule’s topological structure.
- Physiochemical descriptors: Calculated numerical metrics, such as molecular weight, polar surface area, or lipophilicity. [5]
Historically, the industry has relied on tree-based models, like random forests, paired with fingerprints or physiochemical descriptors, and graph-based message passing neural networks. They are robust, quick to train, and computationally inexpensive.
As of late, foundation models have given promising results. These models learn the underlying chemistry by pre-training large deep learning architectures (like Transformers or GNNs) on massive, unlabeled chemical databases using self-supervised learning. We can then fine-tune these models on smaller, project-specific datasets to achieve high predictive accuracy. Ensemble models that combine the achievements of GNNs and classical ML are currently benchmark models for various targets. [6][7]
An essential part of ML workflows is creating meaningful features. If a model is fed poorly engineered features, it will output bad predictive performance; the classic “garbage in, garbage out” setting. Furthermore, there is no universally perfect algorithm; the best model architecture and optimal featurisation strategy depend entirely on what you are researching. Different targets and chemical subspaces behave differently, which requires tailored approaches.
A concrete example: property prediction for compounds
The OpenADMET-ExpansionRx Blind Challenge, closed in early 2026, gave important benchmarks on what type of models and approaches work the best in a practical setting for property prediction. The challenge ended up receiving more than 1000 submissions from over 370 participants, including industrial groups, and has received a lot of community praise for being extremely useful for ML teams focused on biochemistry.
In the competition, participants were tasked to predict nine critical ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) targets for over 7000 molecules from an actual drug discovery campaign. Models were trained on early-stage data and tested on molecules from later stages of the project, which is argued to mimic many of the real-world conditions.
The participating teams implemented everything from classic tree-based algorithms to more complex GNNs and foundation models, and the winning architectures relied mostly on having an ensemble of three to four architecturally distinct models. Furthermore, multitask learning was a clear performance booster in most cases, meaning that training models jointly to learn dependencies between the targets yielded better results compared to independent models, even when the data was sparse.
Usage of external data for pretraining was the key to achieving top performance in this challenge. In the final leaderboard, 4 out of 5 top performers utilised proprietary data in addition to publicly available datasets. Combining large datasets with careful data curation and target-specific engineering was a choice that helped to achieve very high ranks.
OpenADMET has already declared they are committed to hosting blind challenges quarterly, and the next one is already up. [7][8]
What comes in the GenAI era?
Generative AI doesn’t remove the need for predictive models. Every compound a generator proposes is another candidate that has to be filtered, scored, and either pursued or dropped. The bottleneck shifts to deciding which compounds are worth making, and that is still a property prediction problem. Machine learning moves failures earlier in the pipeline, where they are cheaper to absorb.
Featurisation, target-specific tuning, and molecular out-of-distribution evaluation matter at least as much as the architecture of the model. The OpenADMET-ExpansionRx results are a useful reminder that public models don’t generalise as well as their benchmark numbers suggest, and that project-specific work isn’t going away, at least yet.
Large language models are useful in automating text and read heavy tasks, like searching and synthesising information from scientific publications or reading regulatory documents. The physical world is much more complicated than that, and we still need chemists to tell us how the compounds behave. A good example of a background for an ML professional in this field is a combination of a strong understanding of chemistry and data science.
Sources
[1] https://www.orionpharma.com/science/our-scientific-approach/drug-development/
[2] https://www.nebiolab.com/drug-discovery-and-development-process/
[3] Gangwal, A., Ansari, A., Ahmad, I., Azad, A. K., Kumarasamy, V., Subramaniyan, V., & Wong, L. S. (2024). Generative artificial intelligence in drug discovery: basic framework, recent advances, challenges, and opportunities. Frontiers in Pharmacology, 15, 1331062. https://doi.org/10.3389/fphar.2024.1331062
[4] Visan, A. I., & Negut, I. (2024). Integrating artificial intelligence for drug discovery in the context of revolutionizing drug delivery. Life, 14(2), 233. https://doi.org/10.3390/life14020233
[5] Lim, S., Lu, Y., Cho, C. Y., Sung, I., Kim, J., Kim, Y., Park, S., & Kim, S. (2021). A review on compound-protein interaction prediction methods: Data, format, representation and model. Computational and Structural Biotechnology Journal, 19, 1541–1556. https://doi.org/10.1016/j.csbj.2021.03.004
[6] Burns, J., Zalte, A., & Green, W. (2025, June 18). Descriptor-based foundation models for molecular property prediction. arXiv.org. https://arxiv.org/abs/2506.15792v1
[7] https://openadmet.ghost.io/lessons-learned-from-the-openadmet-expansionrx-blind-challenge/
[8] https://huggingface.co/spaces/openadmet/OpenADMET-ExpansionRx-Challenge
Author
Maria Yli-Luukko Senior Data Scientist, Solita